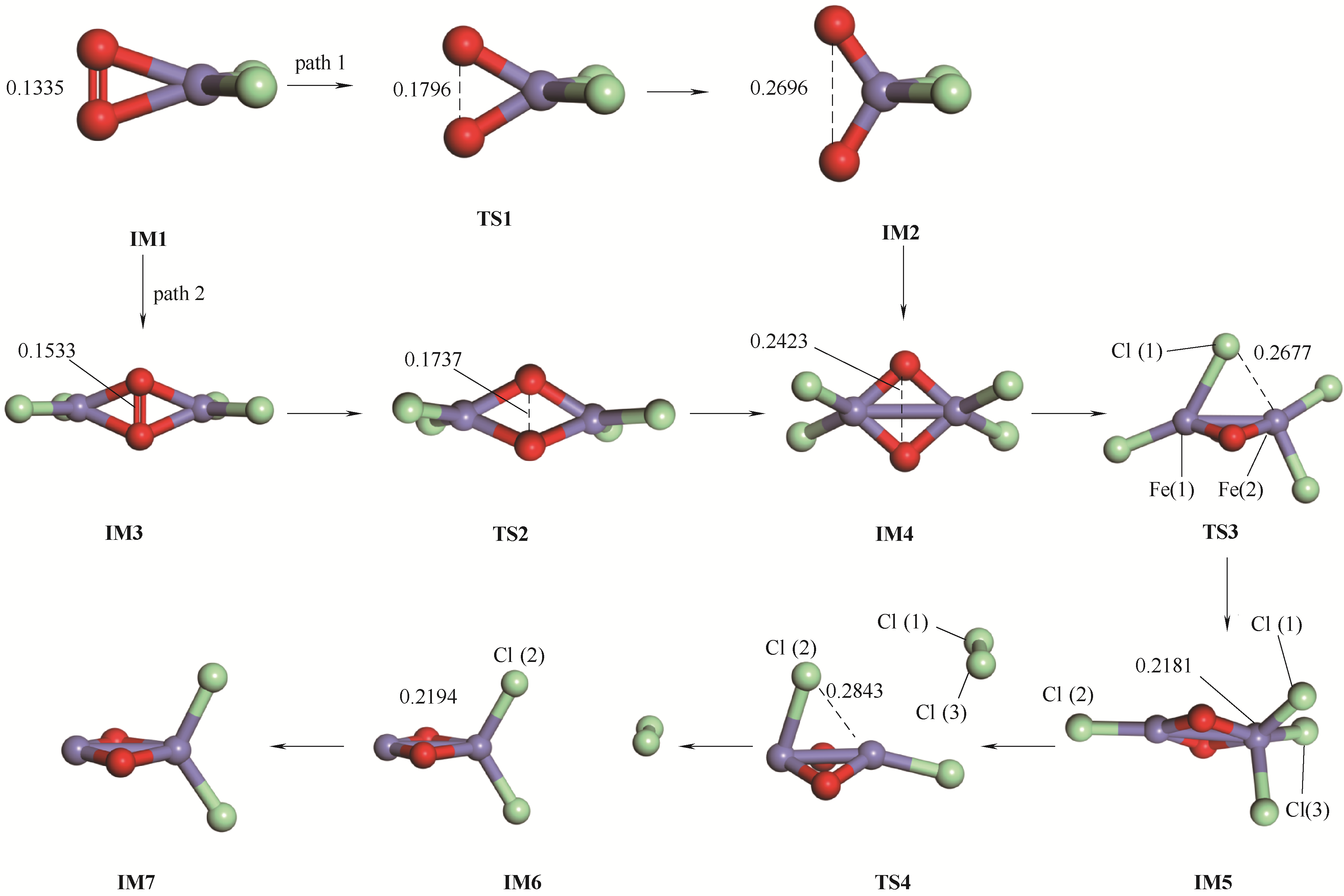
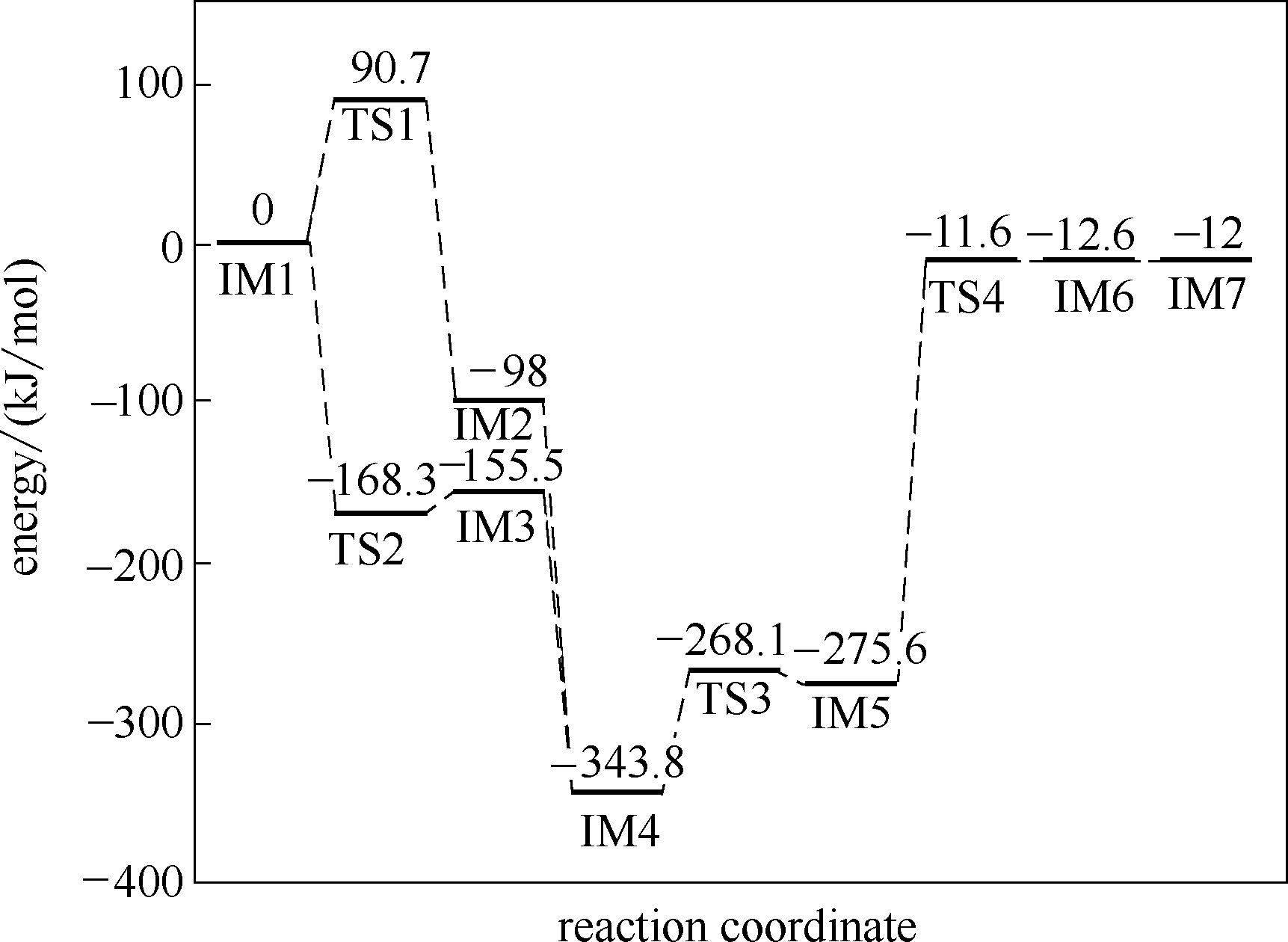
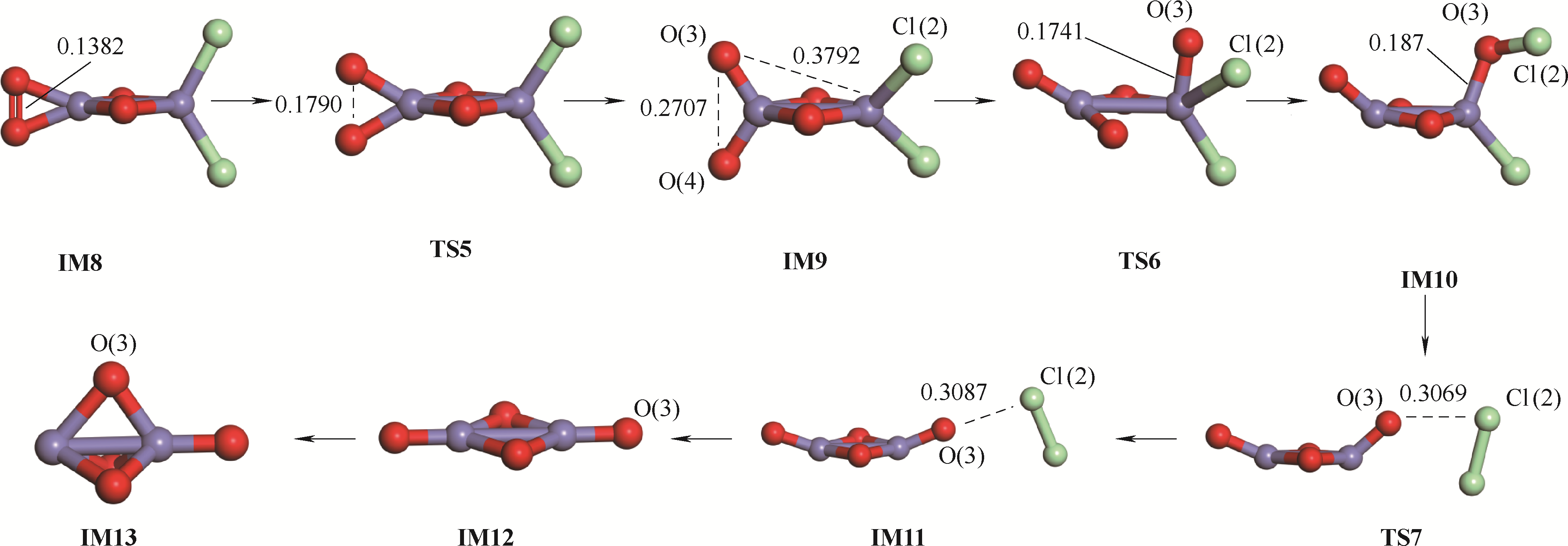
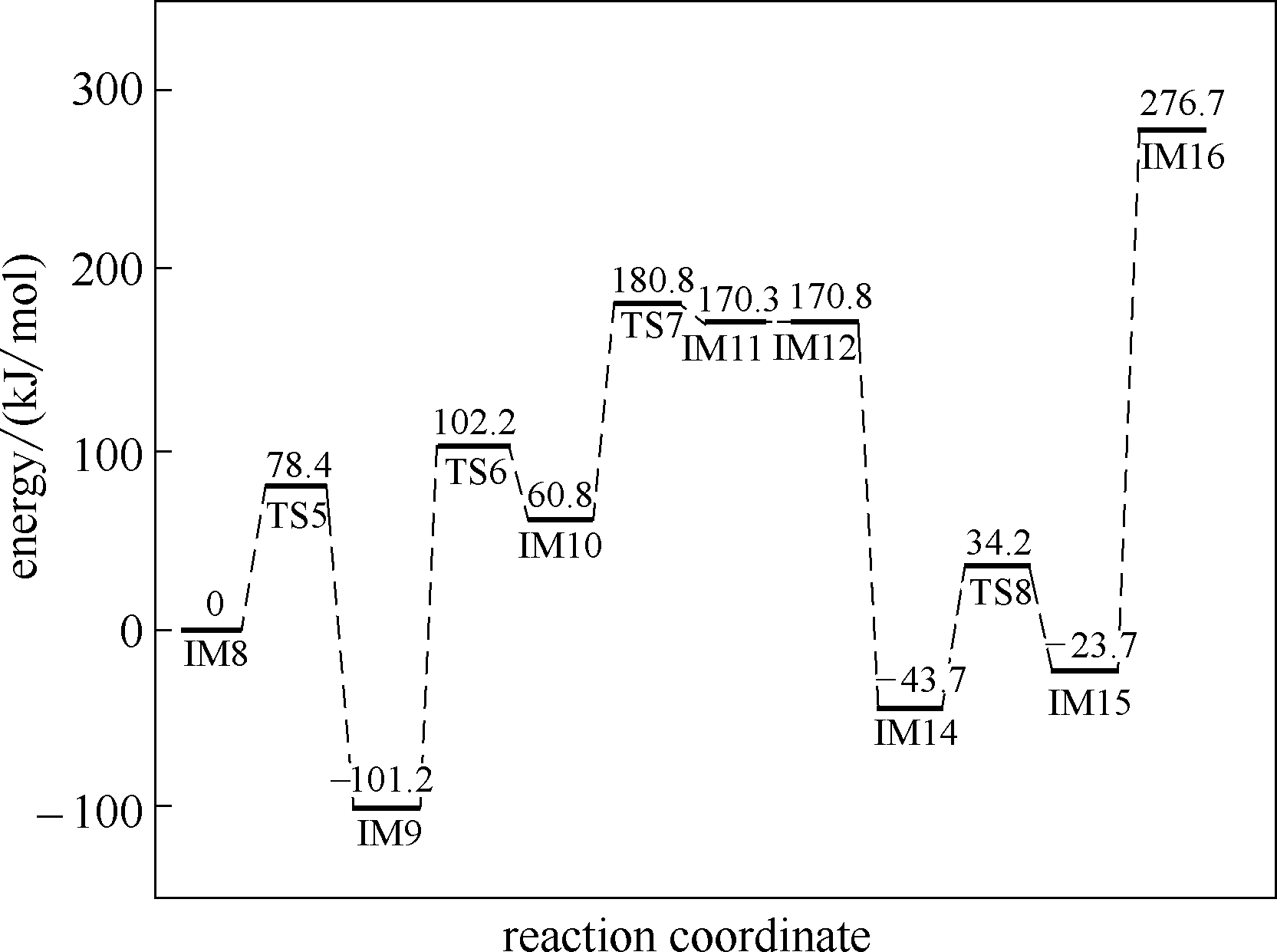
| 1 |
Khan A A , Jong W D , Jansens P J , et al . Biomass combustion in fluidized bed boilers: potential problems and remedies[J]. Fuel Processing Technology, 2009, 90(1): 21-50.
|
| 2 |
Tillman D A . Biomass cofiring: the technology, the experience, the combustion consequences[J]. Biomass & Bioenergy, 2000, 19(6): 365-384.
|
| 3 |
Dincer I . Renewable energy and sustainable development: a crucial review[J]. Renewable and Sustainable Energy Reviews, 2000, 4(2): 157-175.
|
| 4 |
Vassilev S V , Baxter D , Andersen L K , et al . An overview of the chemical composition of biomass[J]. Fuel, 2010, 89(5): 913-933.
|
| 5 |
Yu C , Qin J , Nie H , et al . Experimental research on agglomeration in straw-fired fluidized beds [J]. Applied Energy, 2011, 88(12): 4534-4543.
|
| 6 |
Nielsen H P , Frandsen F J , Dam-Johansen K , et al . The implications of chlorine-associated corrosion on the operation of biomass-fired boilers[J]. Progress in Energy and Combustion Science, 2000, 26(3): 283-298.
|
| 7 |
徐婧 . 生物质燃烧过程中碱金属析出的实验研究[D]. 杭州: 浙江大学, 2006.
|
|
Xu J . Experimental study on alkali metal precipitation during biomass combustion [D]. Hangzhou: Zhejiang University, 2006.
|
| 8 |
Jensen P A , Frandsen F J , Dam-Johansen K , et al . Experimental investigation of the transformation and release to gas phase of potassium and chlorine during straw pyrolysis[J]. Energy&Fuel, 2000, 14(6): 1280-1285.
|
| 9 |
Nielsen H P . The implications of chlorine-associated corrosion on the operation of biomass-fired boilers[J]. Progress in Energy & Combustion Science, 2000, 26(3): 283-298.
|
| 10 |
岳茂振 . 秸秆类生物质与煤混燃过程中氯腐蚀规律研究[D]. 济南: 山东大学, 2011.
|
|
Yue M Z . Study on the corrosion law of chlorine in the process of straw-based biomass and coal co-firing[D]. Jinan: Shandong University, 2011.
|
| 11 |
李至 . 生物质与煤混燃灰熔融特性及其影响研究[D]. 武汉: 华中科技大学, 2015.
|
|
Li Z . Study on melting characteristics and effects of biomass and coal mixed combustion ash [D]. Wuhan: Huazhong University of Science and Technology, 2015.
|
| 12 |
Uusitalo M A , Vuoristo P M J , Mantyla T A . High temperature corrosion of coatings and boiler steels below chlorine-containing salt deposits[J]. Corrosion Science, 2004, 46(6): 1311–1331.
|
| 13 |
Grahke H J . Reese E, Spiegel M. The effect of chlorides, hydrogen chloride, and sulthr dioxide in the oxidation of steels below deposits[J]. Corrosion Science, 1995, 37(7): 1023-1043.
|
| 14 |
Berlanga C , Ruiz J A . Study of corrosion in a biomass boiler[J]. Journal of Chemistry, 2013, 2013(4): 1-8.
|
| 15 |
Zahs A , Spiegel M , Grabke H J . Chloridation and oxidation of iron, chromium, nickel and their alloys in chloridizing and oxidizing atmospheres at 400–700℃[J]. Corrosion Science, 2000, 42(6): 1093-1122.
|
| 16 |
邹潺, 王春波, 邢佳颖 . 煤燃烧过程中砷与氮氧化物的反应机理[J].燃料化学学报, 2019, 47(2): 138-143.
|
|
Zou C , Wang C B , Xing J Y . Reaction mechanism of arsenic and nitrogen oxides in coal combustion process[J]. Journal of Fuel Chemistry and Technology, 2019, 47(2): 138-143.
|
| 17 |
贾建波, 曾凡桂, 李美芬, 等 . 用密度泛函理论研究煤中甲基苯生成甲烷的反应机理[J]. 化工学报, 2010, 61(12): 3235-3242.
|
|
Jia J B , Zeng F G , Li M F , et al . Study on the reaction mechanism of methylbenzene to methane in coal by density functional theory[J]. CIESC Journal, 2010, 61(12): 3235-3242.
|
| 18 |
王荣杰, 沈本贤, 马健, 等 . 基于密度泛函的硫黄S8开环裂解机理[J]. 化工学报, 2015, 66(10): 3919-3924.
|
|
Wang R J , Shen B X , Ma J , et al . Mechanism of open-loop cracking of sulfur S8 based on density functional theory[J]. CIESC Journal, 2015, 66(10): 3919-3924.
|
| 19 |
朱正和, 阎世英, 马美仲 . B2H6分子的几何构型[J]. 物理学报, 2005, 54(7): 3106-3110.
|
|
Zhu Z H , Yan S Y , Ma M Z . Geometrical configuration of B2H6 molecule[J]. Acta Physica Sinica, 2005, 54(7): 3106-3110.
|
| 20 |
冯晓娜, 杨冬花, 武正簧, 等 . B、Al、La 同晶取代 EU-1 分子筛酸性的密度泛函理论计算[J]. 石油学报, 2013, 29(4): 647-654.
|
|
Feng X N , Yang D H , Wu Z H , et al . Density functional theory calculation of the acidity of B, Al and La isomorphous substituted EU-1 molecular sieves[J]. Acta Petrolei Sinica, 2013, 29(4): 647-654.
|
| 21 |
田继权, 王伯初, 陈双扣, 等 . HClO对TYR氧化的理论研究[J]. 计算机与应用化学, 2011, 28(2): 217-221.
|
|
Tian J Q , Wang B C , Chen S K , et al . Theoretical study on oxidation of TYR by HClO[J]. Computers and Applied Chemistry, 2011, 28(2): 217-221.
|
| 22 |
Zhang M , Wang W , Chen Y . Theoretical investigation of selective catalytic reduction of NO on MIL-100-Fe[J]. Physical Chemistry Chemical Physics, 2018, 20(4): 2211-2219.
|
| 23 |
黄金保, 刘朝, 魏顺安 . 纤维素热解形成左旋葡聚糖机理的理论研究[J]. 燃料化学学报, 2011, 39(8): 590-594.
|
|
Huang J B , Liu C , Wei S A . Theoretical study on the mechanism of cellulose pyrolysis to form L-glucan[J]. Journal of Fuel Chemistry and Technology, 2011, 39(8): 590-594.
|
| 24 |
王小曼, 苏亚欣, 周皞, 等 . Fe2与SO2反应的密度泛函理论研究[J]. 原子与分子物理学报, 2017, 34(4): 625-629.
|
|
Wang X M , Su Y X , Zhou H , et al . Density functional theory study on the reaction of Fe2 with SO2 [J]. Journal of Atomic and Molecular Physics, 2017, 34(4): 625-629.
|
| 25 |
侯封, 金晶, 王永贞, 等 . 污泥热解中 HCN 与 CaO 的反应机理: 密度泛函理论研究[J]. 燃料化学学报, 2017, 45(1): 123-128.
|
|
Hou F , Jin J , Wang Y Z , et al . Reaction mechanism of HCN and CaO in sludge pyrolysis: density functional theory study[J]. Journal of Fuel Chemistry and Technology, 2017, 45(1): 123-128.
|
| 26 |
Reddy B V , Rasouli F , Hajaligol M R , et al . Novel pathway for CO oxidation on a Fe2O3 cluster[J]. Chemical Physics Letters, 2004, 384(4/5/6): 242-245.
|
| 27 |
迟绍明, 王宁, 马丽英, 等 . NO3 -+Cl2→ClONO2+Cl-反应势能面和势能阱[J]. 高等学校化学学报, 2008, 29(6): 1228-1233.
|
|
Chi S M , Wang N , Ma L Y , et al . Potential energy surface and potential energy well of NO3 -+Cl2→ClONO2+Cl- [J]. Chemical Journal of Chinese Universities, 2008, 29(6): 1228-1233.
|
| 28 |
Bojana G P , Camaioni D M , Michel D . About the barriers to reaction of CCl4 with HFeOH and FeCl2 [J]. Journal of Physical Chemistry A, 2011, 115(31): 8713-8720.
|
| 29 |
Mejia-Olvera R , Reveles J U , Pacheco-Ortín S M , et al . Molecular dynamics and electronic structure study of neutral, cationic and anionic (Fe3O4) 1–5, clusters[J]. Chemical Physics Letters, 2018, 706(3): 494-500.
|
| 30 |
凌丽霞, 赵俐娟, 章日光, 等 . 苯甲酸和苯甲醛热解机理的量子化学研究[J]. 化工学报, 2009, 60(5): 1224-1230.
|
|
Ling L X , Zhao L J , Zhang R G , et al . Quantum chemistry study on the pyrolysis mechanism of benzoic acid and benzaldehyde[J]. CIESC Journal, 2009, 60(5): 1224-1230.
|
| 31 |
徐建华, 胡常伟 . FeCH2+H2→Fe+CH4反应机理的密度泛函理论研究[J]. 原子与分子物理学报, 2008, 25(2): 274-280.
|
|
Xu J H , Hu C W . Density functional theory study on the reaction mechanism of FeCH2+H2→Fe+CH4 [J]. Journal of Atomic and Molecular Physics, 2008, 25(2): 274-280.
|
| 32 |
Qin W , Chen Q L , Wang Y , et al . Theoretical study of oxidation-reduction reaction of Fe2O3 supported on MgO during chemical looping combustion[J]. Applied Surface Science, 2013, 266(2): 350-354.
|
 ),龙慎伟2,李冠兵2,牛胜利1(
),龙慎伟2,李冠兵2,牛胜利1( 京公网安备 11010102001995号
京公网安备 11010102001995号